Run demultiplexing, the standard method
This describes the anglerfish run mode of running this tool, which expects an input samplesheet specifying the expected Illumina barcodes found in the sequenced ONT flowcell or barcode. Here we discuss some subjects in further details than the overview given in Usage
Use cases
The primary use of anglerfish would be to detect issues in Illumina sequencing pools using a method (ONT sequencing) independent from Illumina. Specific use cases:
When samples are pooled evenly, detect outliers
Detect barcoding issues or potential sample-mixups
And downstream of anglerfish: identify samples by mapping
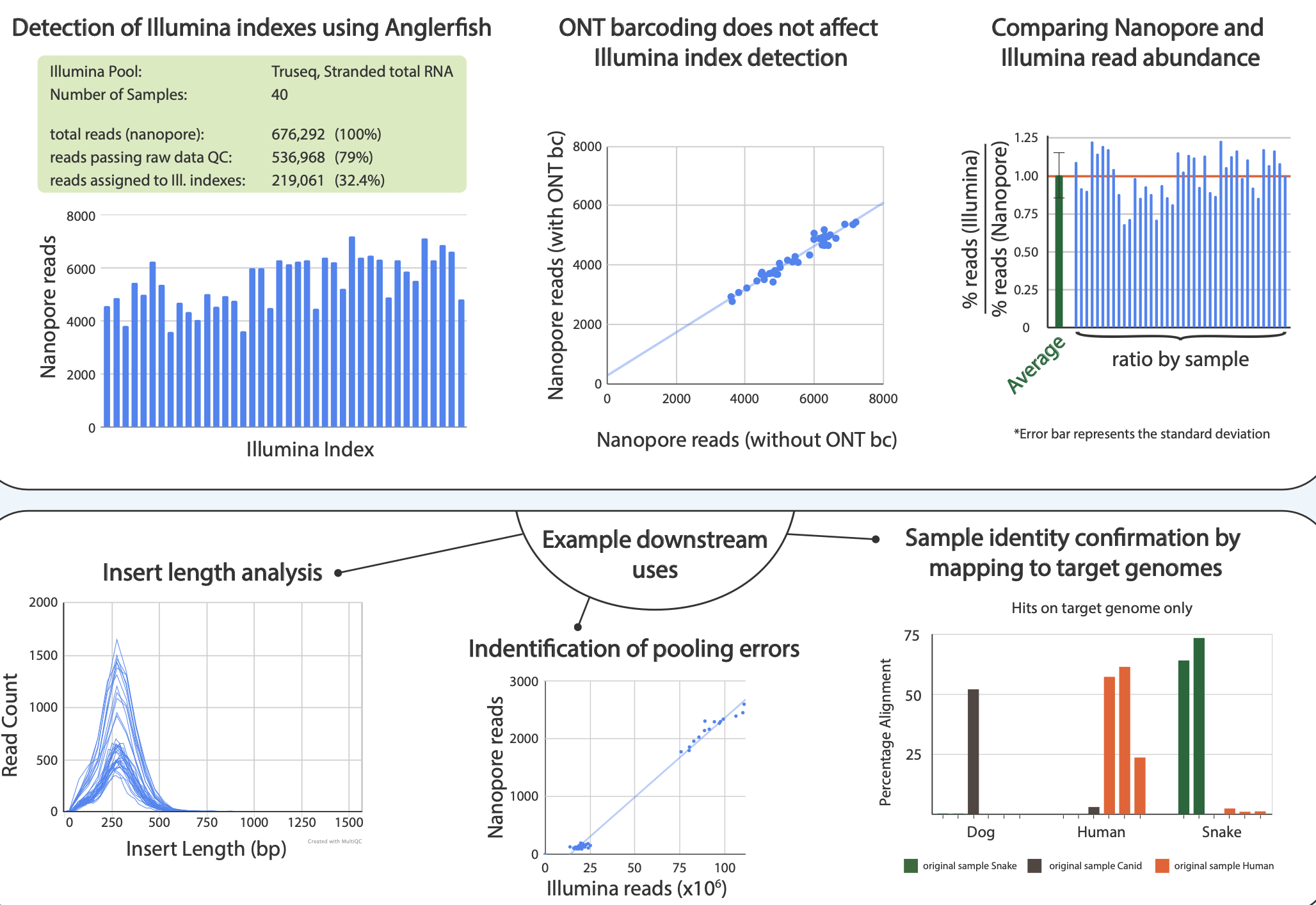
Figure 1. From poster2020. Top row: We compared pool of 40 Illumina barcodes sequenced on a MinION device and demultiplexed using Anglerfish with the outputs from an Illumina sequencer. It shows the abundances are comparable. Bottom row: Downstream uses cases of Anglerfish. Detecting library insert sizes, detecting pooling errors and mapping demultiplexed data to reference genomes.
Output
Example of file output from anglerfish run with a single setup pool (as opposed to a complex one). Without specifying an --out_fastq option, it generates a default name for the output folder.
anglerfish_run_YYYY_MM_DD_HHMMSS
├── anglerfish_dataframe.csv
├── anglerfish_stats.json
├── anglerfish_stats.txt
├── index_len(indexlength).fasta
├── index_len(indexlength).paf
├── sample1.fastq.gz
└── sample2.fastq.gz
The basic operation of Anglerfish is to map the input reads to a template of the adaptors index_len(indexlength).fasta using
minimap2 - output as an alignment file to index_len(indexlength).paf.
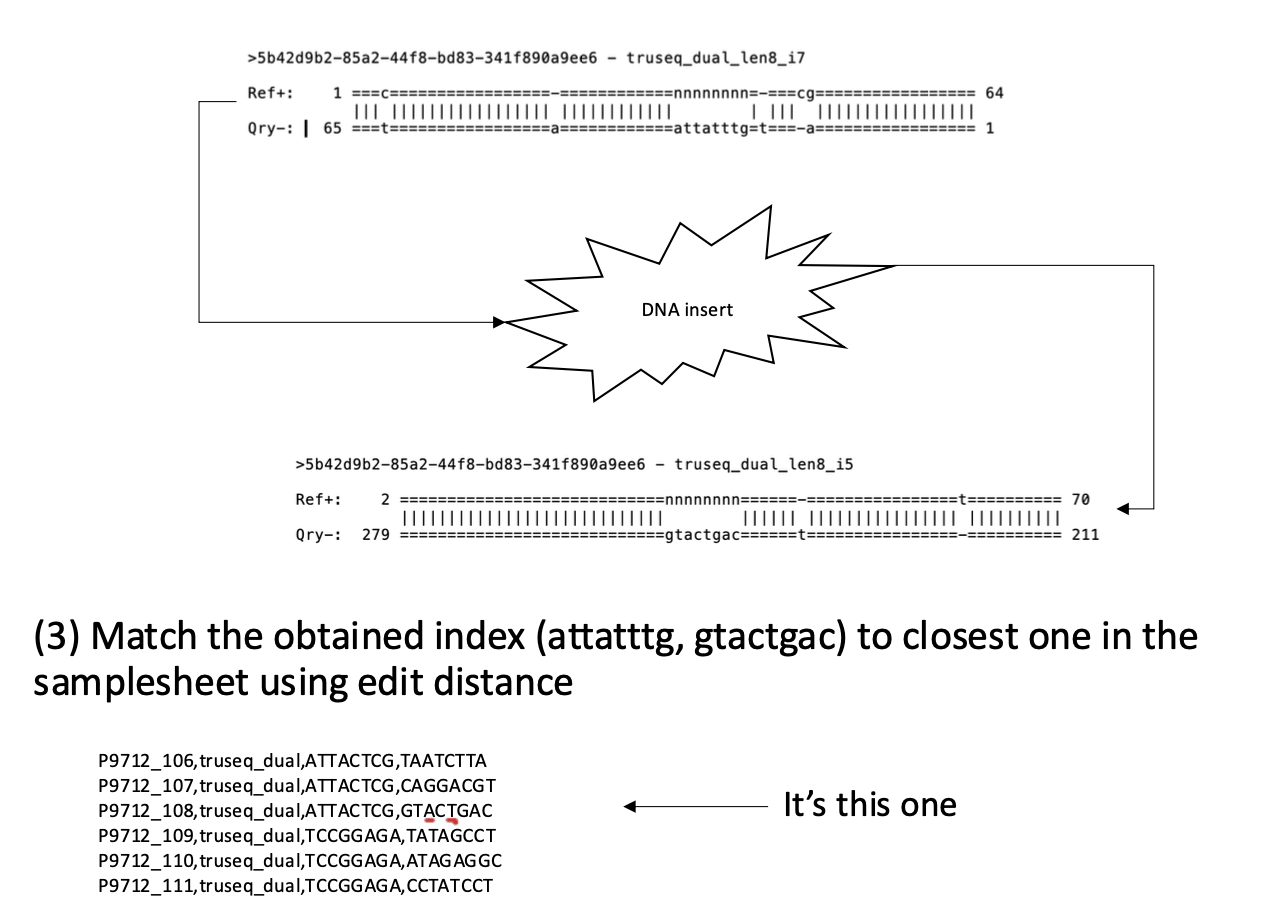
Figure 2. An example of how read mapping in Anglerfish works. Adapter templates for I7 and I5 map to the ends of the read “5b42”, the Illumina barcodes are read from the “N” gap in the templates.
Anglerfish reports the stats of the run to a report called anglerfish_stats.txt, with the same number found in a machine readable JSON format (anglerfish_stats.json). Let’s look at a few fields from this report and number the lines:
01: Anglerfish v. 0.7.0 (run: anglerfish_2024_10_28_153312, 5c98ad62-784d-4b27-8dd4-a69bbfe553ac)
02: ===================
03: truseq_dual:
04: 105608 input_reads (100.00%)
05: 96593 reads aligning to adaptor sequences (91.46%)
06: 54785 aligned reads matching both I7 and I5 adaptor (56.72%)
07: 33658 aligned reads matching only I7 or I5 adaptor (34.85%)
08: 492 aligned reads matching multiple I7/I5 adaptor pairs (0.51%)
09: 7658 aligned reads with uncategorized alignments (7.93%)
03: Each adapter type will have their own section in the header
04: Any alignment from minimap given constraints of the parameters it’s given
06: Reads matching the template (even partially) adapter1-insert-adapter2
08-09: Any reads falling outside of the adapter1-insert-adapter2 expectation. One reason for this could be incomplete splitting of chimeric reads by the sequencing software. Anglerfish will not resolve such reads, and these cases have not been studied by the anglerfish authors.
The anglerfish_dataframe.csv file summarizes all index level stats (samplesheet samples and unknown indexes) into a single “flat” table.
And finally, the DNA inserts of each demultiplexed read will be output into fastq files according the samplesheet in sample1.fastq.gz, sample2.fastq.gz, etc.
Mixed setup pools
Anglerfish supports demultiplexing complex Illumina pools containing a mix of adapter setups, e.g. mixing samples with different index types like “truseq” types and “nextera” and samples with different lengths like 8+8bp and 6bp (single index). See this reference from Illumina to get an idea of how these indexes might differ.
Example of such a samplesheet:
dual1,truseq_dual,TAATGCGC-CAGGACGT,/path/to/ONTreads.fastq.gz
dual2,truseq_dual,TAATGCGC-GTACTGAC,/path/to/ONTreads.fastq.gz
dual3,truseq_dual,ATTACTCG-TATAGCCT,/path/to/ONTreads.fastq.gz
single1,truseq,GAAACCCT,/path/to/ONTreads.fastq.gz
single2,truseq,CTGACTGA,/path/to/ONTreads.fastq.gz
single3,truseq,TCTCAGTG,/path/to/ONTreads.fastq.gz
The way these are handled are, for each adapter-type and index length combination present, separate minimap runs and read clustering is performed, then the results are aggregated in the report.
The path the fastq files supports glob’ing, e.g. you can specify multiple files like /path/to/flowcell/fastq_passed/*.fastq.gz
Multiple ONT barcodes
Anglerfish has support for ONT barcoding using the option -n, --ont_barcodes. It does however assume a directory structure that is set by the sequencing software MinKNOW, where the fastq files of the demultiplexed ONT barcodes are arranged into folders name barcode01, barcode02, …, e.g.:
20250207_1125_1F_NNN12345_fa78ca0f/
├── barcode01
├── barcode02
├── barcode03
└── barcode04
Let’s say barcode01 and barcode02 contain Illumina pools you are interested in demultiplexing using anglerfish. The samplesheet you give might look something like this:
dual1,truseq_dual,TAATGCGC-CAGGACGT,/path/to/20250207_1125_1F_NNN12345_fa78ca0f/barcode01/*.fastq.gz
dual2,truseq_dual,TAATGCGC-GTACTGAC,/path/to/20250207_1125_1F_NNN12345_fa78ca0f/barcode01/*.fastq.gz
single1,truseq,GAAACCCT,/path/to/20250207_1125_1F_NNN12345_fa78ca0f/barcode02/*.fastq.gz
single2,truseq,CTGACTGA,/path/to/20250207_1125_1F_NNN12345_fa78ca0f/barcode02/*.fastq.gz
Unknown indexes
Indexes that do not match (within a set edit distance) the indexes in the samplesheet will be listed in descending order at the bottom of the report.
These unknown matches are not clustered by sequence, such that each read error will get its own entry therefore the list is truncated at # samples in samplesheet + 10. The column closest_match lists the samples(s) which have the shortest edit distance to this sequence.
The results might be distorted when the input fastq file(s) contain a mixed adapter setup. For the samplesheet given above, the unknown index list of both groups of adapter-index groups will be combined in the report. So the single-index group will contain hits to the “real”, known dual index indices. E.g. “TAATGCGC” from “dual1” and “dual2” will be listed.
Lenient mode
A common category of human error when inputting samplesheets for sequencing instruments are mixing up the orientations of the barcodes, e.g. erroneously reverse complementing of the entire column of i5 indices. This is not helped by Illumina changing standards in their versions of samplesheets.
To correct for this error in Anglerfish and still be able to evaluate other pooling issues at the same time, an extra mode is available called lenient (with option -l, --lenient). Essentially this mode will demultiplex the samplesheet four times using all the possible orientations of the indexes, i.e. all samples normal-I5 + normal-I7, all samples reverse comp-I5 + normal-I7, ….
If one of the four runs yield 4 times more (adjustable using the option -x, --lenient_factor ) demultiplexed reads than the second most abundant run - it will be the one reported in the anglerfish_stats.txt report.
Additionally you can force the anglerfish to run in one of these three alternative index orientations using the -p, --force_rc [i7|i5|i7+i5|original], this will however disable lenient mode.